Tagged PDF documents with forms
This technique relates to:
See PDF Technology Notes for information on user agent and assistive technology support.
The objective of this technique is to notify the user when a field that must be completed has not been completed in a PDF form. Required fields are implemented using the /Ff entry in the form field's dictionary (see Table 220 in Section 12.7 (Interactive Forms) of PDF 1.7 (ISO 32000-1). This is normally accomplished using a tool for authoring PDF.
If errors are found, an alert dialog describes the nature of the error in text. This may be accomplished through scripting created by the author (see, for example, SCR18: Providing client-side validation and alert). User agents, such as Adobe Acrobat Pro and LiveCycle, can provide automatic alerts (as described in the examples below).
Note: once the user dismisses the alert dialog, it may be helpful if the script positions the keyboard focus on the field where the error occurred, although some users may expect the focus to remain on the last control focused prior to the alert appearing. Authors should exercise care to ensure that any movement of the focus will be expected. For example, if the alert announces a missing required phone number, positioning the focus on the phone number field when the alert is dismissed can be regarded as helpful and expected. In some cases, however, this may not be possible. If multiple input errors occur on the page, another approach must be taken to error reporting. (See, for example, the Adobe scripting resources.)
Ensuring that users are aware an error has occurred, can determine what is wrong, and can correct it are keys to software usability and accessibility. Meeting this objective helps ensure that all users can complete transactions with ease and confidence.
It is also important that users are aware that an error may occur. You can incorporate this information in labels; for example, "Date (required)" or the use of a red asterisk to indicate required fields. (Make sure that a legend appears on each form with required fields, e.g., "* = required field".) See PDF10: Providing labels for interactive form controls in PDF documents.
This example is shown with Adobe Acrobat Pro. There are other software tools that perform similar functions. See the list of other software tools in PDF Authoring Tools that Provide Accessibility Support.
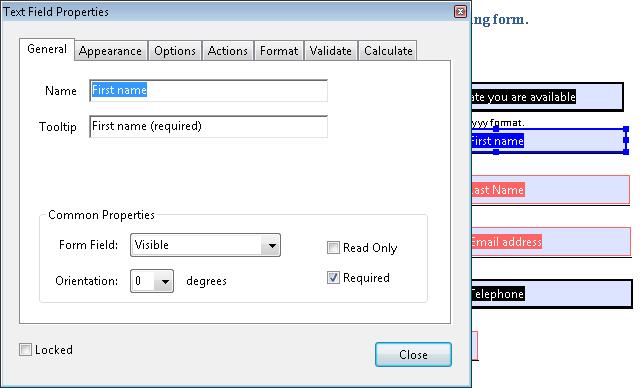
Access the context menu of the field and select the Properties dialog.
If the field is required, check the Required box. This checkbox forces the user to fill in the selected form field. If the user attempts to submit the form while a required field is blank, an error message appears and the empty required form field is highlighted.
This example is shown in operation in the working example of creating a required field in Acrobat.
This example is shown with Adobe LiveCycle Designer. There are other software tools that perform similar functions. See the list of other software tools in PDF Authoring Tools that Provide Accessibility Support.
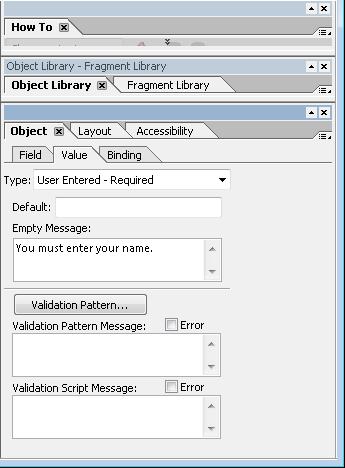
Access the context menu of the form control, select Palettes, and select Object.
Select "User entered - Required" from the Type pulldown.
Enter an error message in the "Empty Message" field. This message appears when a user tries to submit the form without entering a value in the required field.
If the user attempts to submit the form with a required field left blank, the Empty Message text appears and the empty required field is highlighted.
The image below shows the Adobe LiveCycle Object palette with the required selection.
You can also add explicit text to the form label to indicate required fields (e.g., "(Required)").
This example is shown in operation in the working example of creating a required field in LiveCycle Designer.
The following code fragment illustrates code that is typical for the object definitions for a typical text field. Note that the text field is required, using the Ff flag. This is typically accomplished by an authoring tool.
<< /AP -dict-
/DA /Helv 0 Tf 0 g
/DR -dict-
/F 0x4
/FT Tx % FT key set to Tx for Text Field
/Ff 0x2 % Ff integer 0x2 value indicates required
/P -dict-
/Rect -array-
/StructParent 0x1
/Subtype Widget
/T First % Partial field name First
/TU First name (required) % TU tool tip value serves as short description
/Type Annot
/V Pat Jones
>>
...
<Start Stream>
BT
/P <</MCID 0 >>BDC
/CS0 cs 0 scn
/TT0 1 Tf
-0.001 Tc 0.003 Tw 11.04 0 0 11.04 72 709.56 Tm
[(P)-6(le)-3(as)10(e)-3( )11(P)-6(rin)2(t)-3( Y)8(o)-7(u)2(r N)4(a)11(m)-6(e)]TJ
0 Tc 0 Tw 9.533 0 Td
( )Tj
-0.004 Tc 0.004 Tw 0.217 0 Td
[(\()-5(R)-4(e)5(q)-1(u)-1(i)-3(r)-3(e)-6(d)-1(\))]TJ
EMC
/P <</MCID 1 >>BDC
0 Tc 0 Tw 4.283 0 Td
[( )-2( )]TJ
EMC
/ArtifactSpan <</MCID 2 >>BDC
0.002 Tc -0.002 Tw 0.456 0 Td
[(__)11(___)11(___)11(___)11(___)11(_)11(____)11(___)11(___)11(__)]TJ
0 Tc 0 Tw 13.391 0 Td
( )Tj
EMC
ET
<End Stream>
Resources are for information purposes only, no endorsement implied.
Section 12.7 (Interactive Forms) in PDF 1.7 (ISO 32000-1)
For each form field that is required, verify that validation information and instructions are provided by applying the following:
Check that the required status is indicated in the form control's label.
Leave the field blank and submit the form. Check that an alert describing the error is provided.
Use a tool that exposes the document through the accessibility API, and verify that the required property is indicated.
#1, #2, and #3 are true.
If this is a sufficient technique for a success criterion, failing this test procedure does not necessarily mean that the success criterion has not been satisfied in some other way, only that this technique has not been successfully implemented and can not be used to claim conformance.
Techniques are informative—that means they are not required. The basis for determining conformance to WCAG 2.0 is the success criteria from the WCAG 2.0 standard—not the techniques. For important information about techniques, please see the Understanding Techniques for WCAG Success Criteria section of Understanding WCAG 2.0.